

Latest kidney cancer news

19 Apr 2024
Sick pay - cancer patients are counting the cost

17 Apr 2024
Cancer52 manifesto launch
12 Apr 2024
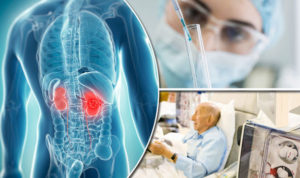
Immunotherapy combinations versus TKIs for papillary renal cell carcinoma
9 Apr 2024
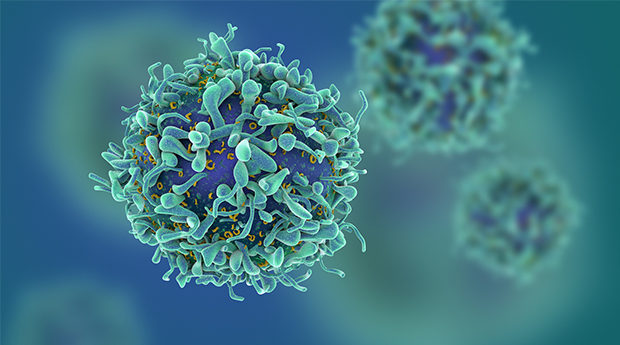
CAR T cell immunotherapy shows promise for patients with kidney cancer
9 Apr 2024
Real world data on the use of cabozantinib plus immunotherapy in the second line

25 Mar 2024
How to manage anxiety and depression when you have kidney cancer

12 Mar 2024
Immunotherapy improves survival in kidney cancer patients with brain metastases
8 Mar 2024
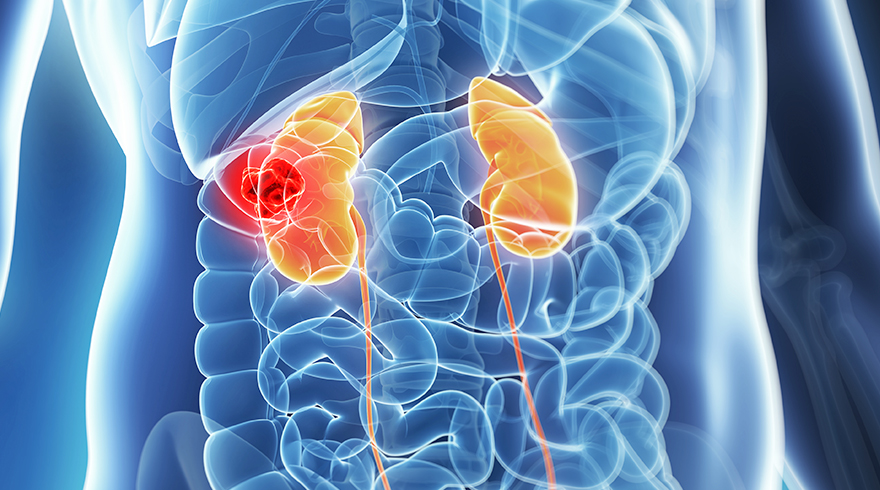
Radiotherapy for the management of renal medullary carcinoma

8 Mar 2024
Manchester patient focus group

8 Mar 2024
NICE approves cabozantinib plus nivolumab for advanced kidney cancer
15 Feb 2024
PRISM study highlights possibility of reducing dose of immunotherapy

15 Feb 2024
World-class radiotherapy in the UK
Action Kidney Cancer
Friendship...community...encouragement...advocacy...information...support...kindness...advice...shared experiences...patient stories...research news...expert insights...empowerment...





Cancer52 manifesto launch
Yesterday (16 April 2024) Cancer52, an alliance of over 100 charities representing rare and less common cancers, launched The Other Half. A manifesto to transform outcomes for people with rare and less common cancers. The event was hosted by Will Quince MP at the House of Commons, Westminster. Actio...


[php_everywhere instance="3"]